Our research
Genome Organization
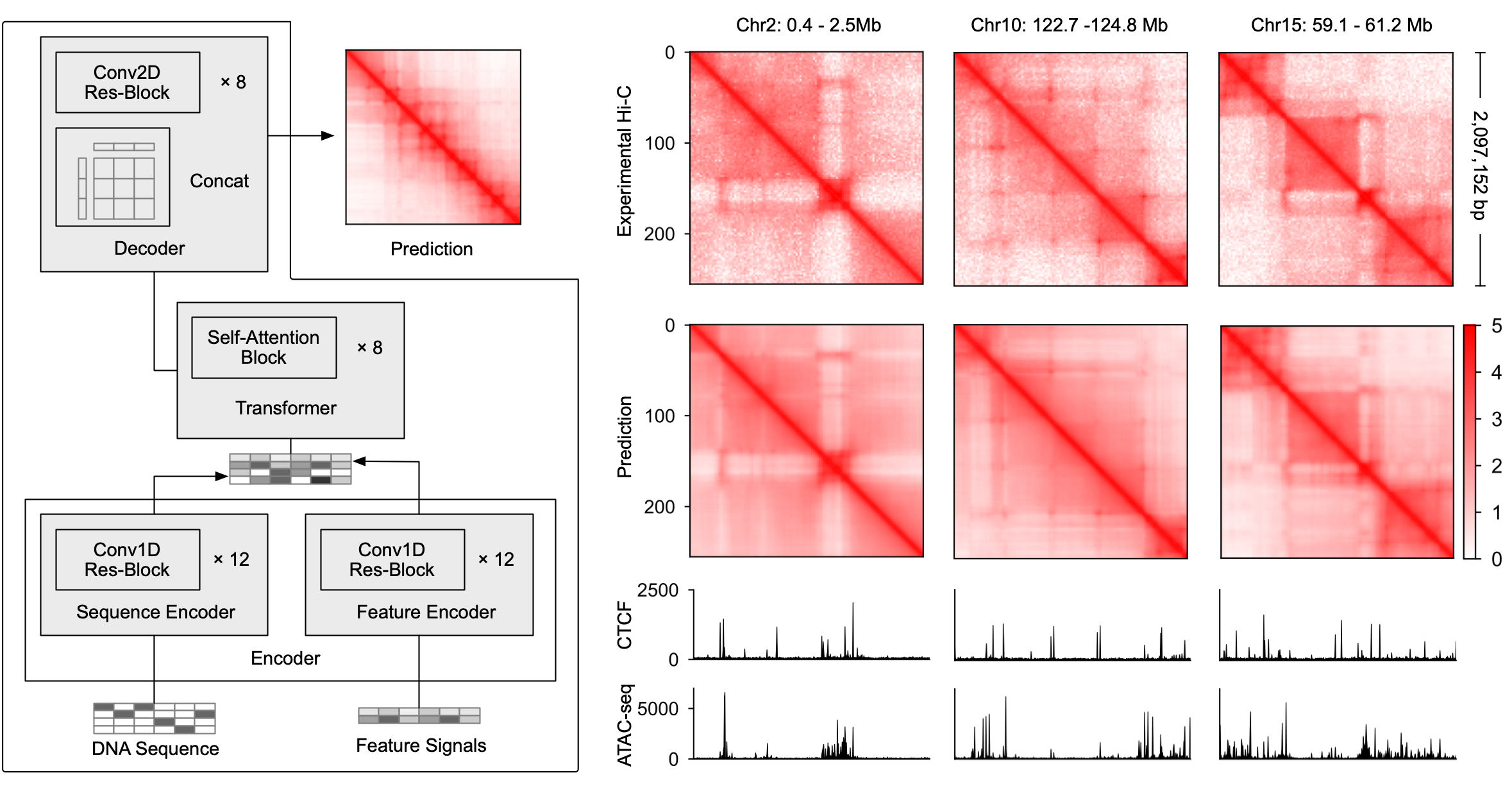
The three-dimensional organization of the genome plays a central role in determining how genes are regulated across cell types, but measuring genome folding experimentally remains costly and difficult to scale. We developed C.Origami, a multimodal deep learning model that predicts cell-type-specific chromatin organization from DNA sequence, chromatin accessibility, and CTCF binding (Tan et al. Nat. Biotechnol. 2023). Through accurate in silico prediction of chromatin interactions, C.Origami allows us to computationally test how regulatory elements alter genome folding. We further developed in silico genetic screening approaches to identify DNA elements and transcriptional regulators that shape chromatin architecture in normal and disease contexts. (Publication, code, model weights, colab demo)
DNA-binding Protein
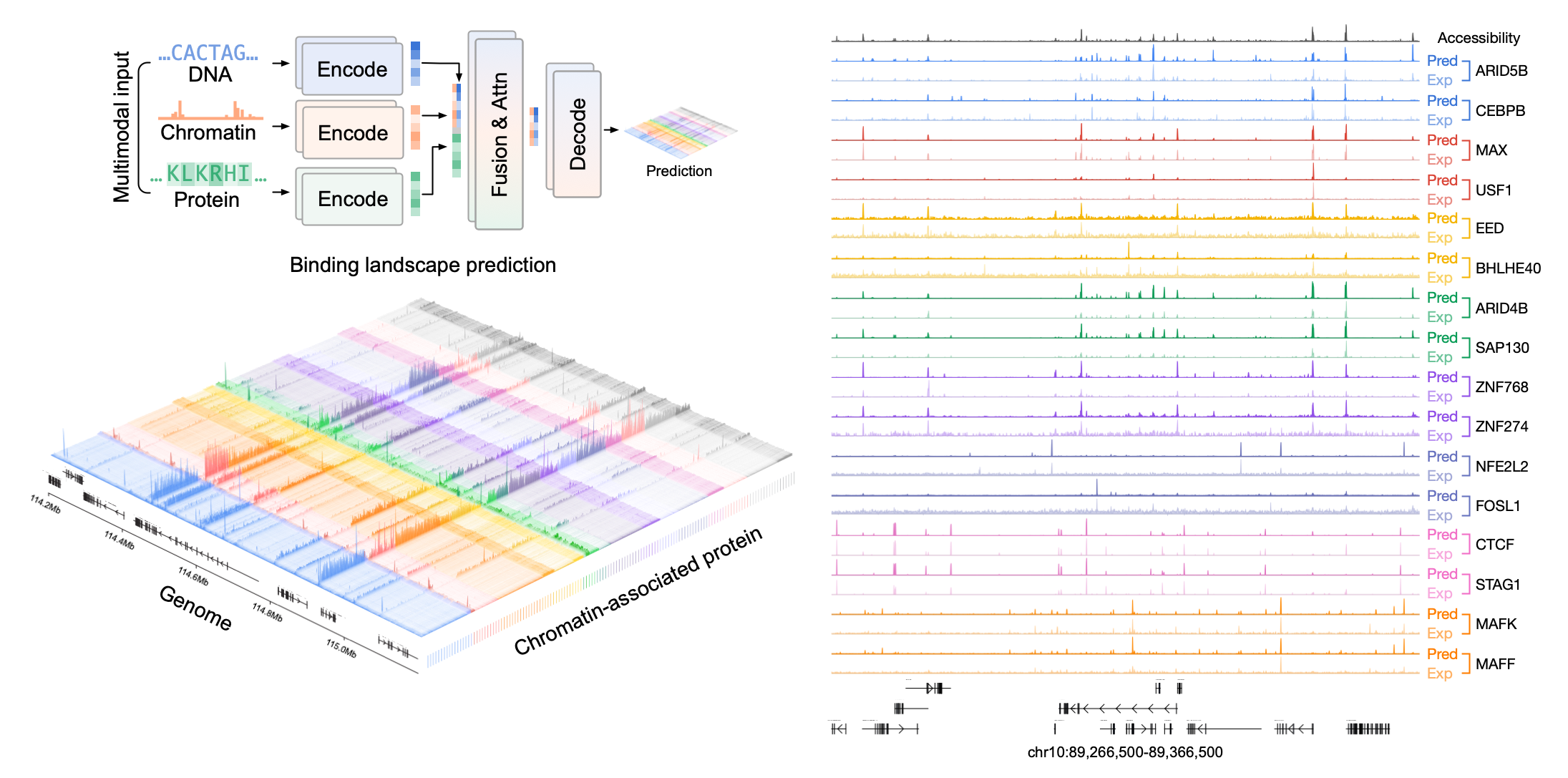
Cell identity is controlled by thousands of regulatory proteins that bind to the genome and coordinate gene expression programs. However, experimentally mapping the binding profiles of these proteins across diverse cell types, disease states, and developmental trajectories remains a major challenge. We developed Chromnitron, a multimodal foundation model that learns the rules of protein–DNA interaction by integrating DNA sequence, chromatin accessibility, and protein amino acid sequence (Tan et al. bioRxiv 2025). Chromnitron can predict cell-type-specific binding landscapes for regulatory proteins, including proteins and cell types not seen during training. Using single-cell chromatin accessibility data, the model can reconstruct dynamic regulatory programs across cell states and identify candidate regulatory proteins that drive immune function, development, and disease. (Preprint, code, model weights, colab demo)
Cellular Niche
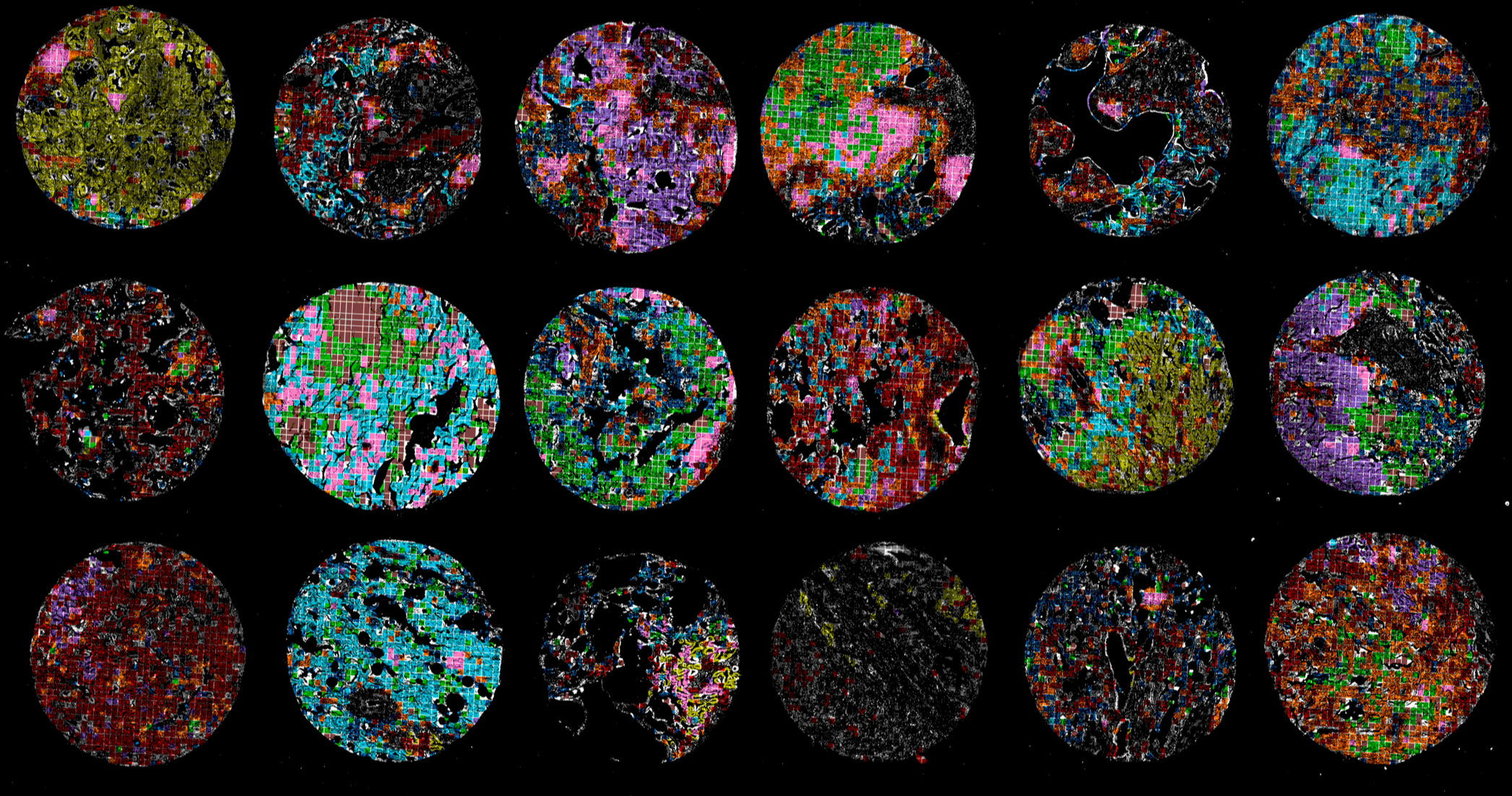
In tissues and tumors, cells are organized into spatial niches that shape immune responses, disease progression, and therapeutic resistance. Understanding these cellular neighborhoods is essential for improving diagnosis, patient stratification, and precision therapy. We developed CANVAS, a self-supervised representation learning framework for analyzing highly multiplexed imaging data (Tan et al. Nat. Biomed. Eng. 2025). Unlike traditional approaches that rely on cell segmentation, CANVAS learns directly from pixel-level spatial images, preserving local morphology, biomarker distributions, and tissue architecture. Applying CANVAS to tumor imaging data, we identified microenvironment signatures that distinguish tumor niches and stratify patients into clinically relevant subgroups. (Publication, code, sample data, colab demo)